A subscription to JoVE is required to view this content. Sign in or start your free trial.
Single-Particle Interferometric Reflectance Imaging Characterization of Individual Extracellular Vesicles and Population Dynamics
In This Article
Summary
This protocol presents single-particle interferometric reflectance imaging that is designed for the multi-level and comprehensive measurements of extracellular vesicles (EV) size, EV count, EV phenotype, and EV biomarker colocalization.
Abstract
Extracellular vesicles (EVs) are nanometer-sized vesicles with a lipid bilayer that are secreted by most cells. EVs carry a multitude of different biological molecules, including protein, lipid, DNA, and RNA, and are postulated to facilitate cell-to-cell communication in diverse tissues and organs. Recently, EVs have attracted significant attention as biomarkers for diagnostics and therapeutic agents for various diseases. Many methods have been developed for EV characterization. However, current methods for EV analysis all have different limitations. Thus, developing efficient and effective methods for EV isolation and characterization remains one of the crucial steps for this cutting-edge research field as it matures. Here, we provide a detailed protocol outlining a single-particle interferometric reflectance imaging sensor (SP-IRIS), as a method that is capable of detecting and characterizing EVs from unpurified biological sources and purified EVs by other methodologies. This advanced technique can be used for multi-level and comprehensive measurements for the analysis of EV size, EV count, EV phenotype, and biomarker colocalization.
Introduction
Extracellular vesicles (EVs) are nanometer-sized membrane vesicles of cellular origin that can be isolated from numerous biological fluids, including blood, breast milk, saliva, urine, bile, pancreatic juice, and cerebrospinal and peritoneal fluids. Derivation of EVs occurs via three main mechanisms: apoptosis, release via fusion of multivesicular bodies with the plasma membrane, and blebbing of the plasma membrane1. Evidence for EV transfer of donor cell components to neighboring or distant cells and tissues suggests these membrane enclosed packages may play important roles in paracrine as well as long distance or endocrine signaling cascades1,2,3. Because EVs can provide a snapshot of a cell's phenotype, the potential for their use as diagnostic and therapeutic tools for the treatment of various diseases has become an active area of research4,5,6,7,8.
Many methods aimed at EV characterization have been developed9,10,11,12,13. Most of these methods provide unique and valuable information about populations of EVs primarily in bulk. While a subset of these techniques can provide details regarding substances within or on single EVs, there can be limitations to characterizing EVs at the single EV level. For example, immuno-electron microscopy can be used to understand single EVs and their composition, but this technique is low throughput, severely limited in its ability to be used for describing population dynamics, and requires significant methods development14.
Recently, development and commercialization of the single-particle interferometric reflectance imaging sensor (SP-IRIS) technique, via the ExoView platform, has opened individual EV characterization using a routine and simple automated data collection method. The core of this technology is the chip, a 1 cm x 1 cm Si/SiO2 double layer, which enables the interferometric measurement of single biological nanoparticles. The chip is tilled with a microarray of individual functionalized antibody spots, allowing for multiplexed detection of up to six different capture types. Standard chips include the common tetraspanin markers (CD81, CD63, and CD9) for capture during the incubation step, and the user can add additional custom capture spots to isolate distinct populations of EVs separate from the tetraspanins. After the incubation step, each capture spot has bound many EVs to it which express the corresponding marker. These captured EVs can then be simply washed, dried, and scanned in the reader to quantify the size of vesicles bound to the capture spot between 50-200 nm to give a number weighted size distribution via SP-IRIS15. The system also offers three fluorescent detection channels for immunolabeling the captured EVs, and provides both the mean fluorescent intensity, which is not limited by the size such as SP-IRIS measurements, and colocalization aspects for each fluorescent stain. This allows the user to define populations of single EVs based on the display of four different biomarkers per EV (capture plus three immunofluorescent labels). The system can go beyond measuring surface proteins with immunofluorescence, as an optional cargo protocol allows the user to probe for interior proteins of the captured EVs and luminal epitopes of membrane spanning surface markers, as well as allows the user to check for EV membrane integrity. In this article, we provide a detailed protocol outlining the steps necessary to obtain consistent data regarding EV size and number, with up to four different biomarkers at a single EV level on large populations of EVs. This technique can be used on both unprocessed biological fluids and EVs isolated using any number of techniques, such as ultracentrifugation, ultrafiltration, precipitating agents, immunoaffinity capture, microfluidics, and size-exclusion chromatography.
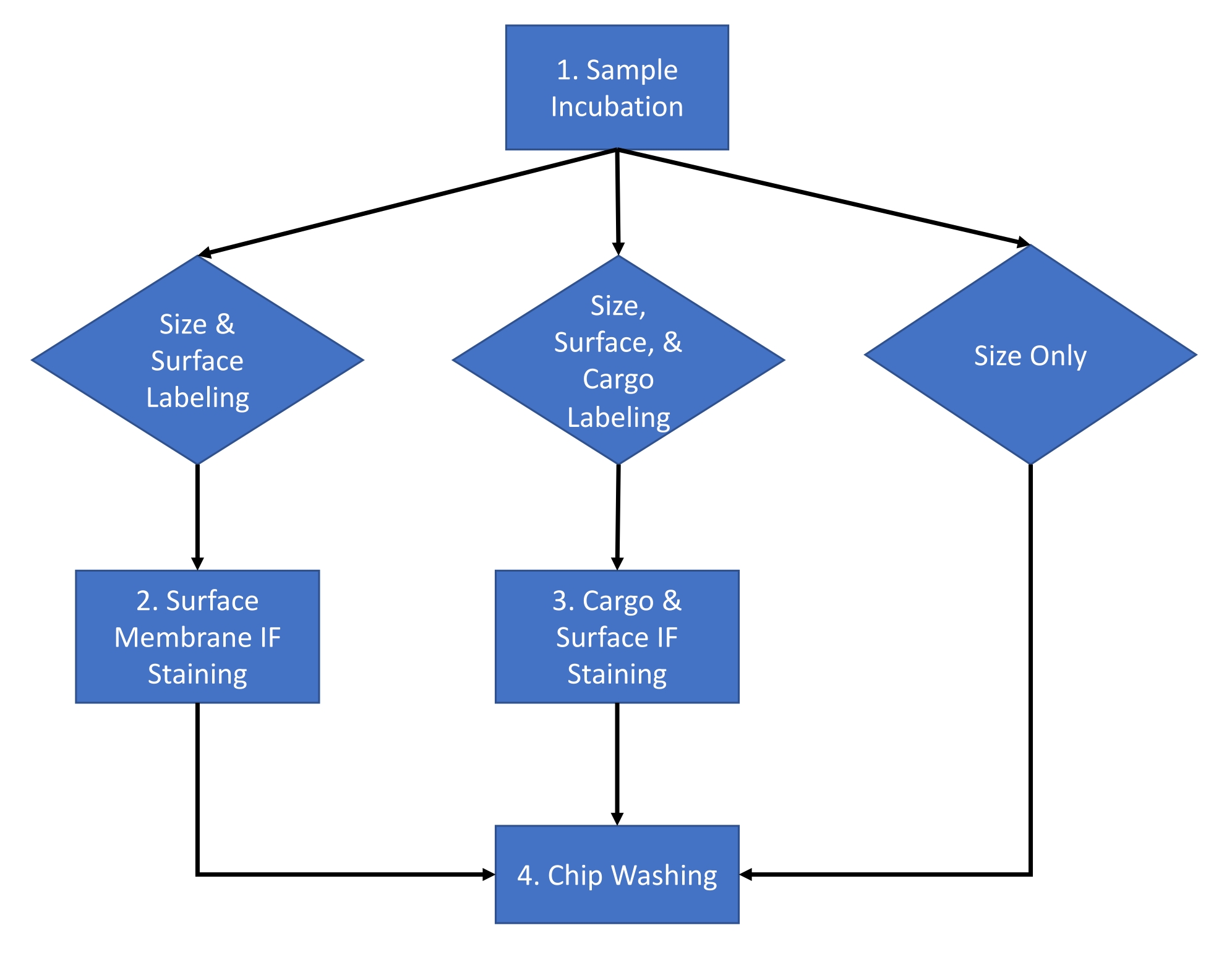
The protocol described below uses extracellular vesicles (EV) derived from HEK 293 cell culture media and from the mouse serum using an established isolation method16. The protocol has been applied to numerous other biological fluids, cell culture medium, and purified extracellular vesicles isolated from biological fluids. This protocol is divided into a two-day procedure with the workflow for a typical experiment shown in Figure 1.

Figure 1: Assay workflow. Assay workflow for choosing the type of analysis to be completed for the sample between size and count, size count and surface staining, and size count and cargo staining. Please click here to view a larger version of this figure.
{kind=link}
Protocol
Serum samples were collected from mice according to an approved Institutional Animal Care and Use Committees (IACUC) at the University of Kansas Medical Center (KUMC) protocol. Use of these biological samples in these experiments was also approved by KUMC.
1. Sample preparation (Day 1)
- Determine EV concentration using nanoparticle tracking or an equivalent technique.
- Dilute the sample with the incubation solution to a concentration of 5 x 107-5 x 108 EVs/mL; a minimum of 50 µL is needed.
NOTE: For samples in which the EV concentration is unknown, total protein at 1 µg/mL can be used as a substitute measure. If the sample is expected to be low in concentration, perform at least a 1:1 dilution in the Incubation Solution before loading. - Place a 24-well plate on a flat surface, free of vibrations and brusque movements.
NOTE: To enhance contrast, a white sheet of paper can be placed under the plate. - Add water to the areas surrounding the wells (Figure 2).

Figure 2: 24-well plate layout. The locations of where to aliquot ddH2O (blue dye was added for visualization purposes only) and the wells in which the chips will be held is shown. Please click here to view a larger version of this figure.
{kind=link}
2. Preparing and prescanning chips
- Remove the sealed 48-well plate containing chips from the 4-8 °C fridge and allow them to reach to room temperature (~15 min) before opening the seal.
NOTE: This is essential to avoid condensation on the chips which can damage the spots. If condensation is observed on the surface upon removing the chip from the pack, wait longer before removing others. - Proceed to step 8 to retrieve chuck to prepare for the pre-scan run (Figure 3).
NOTE: The pre-scan data will be used to identify any particles that are detectable on the capture spots prior to the incubation with the sample so they can be removed during the analysis step.

Figure 3: Image of the chuck used to load the chip into the machine. Please click here to view a larger version of this figure.
{kind=link}
- Using tweezers, remove the desired number of chips (one chip per sample) from the 48-well plate and load the chips directly into the chuck for the prescan run. When all of the chips intended to be used for the experiment have been loaded and prescanned as described in step 8, proceed to step 2.4.
NOTE: When handling chips, be sure to not touch the squares in the center, as the antibody capture spots are in this region and will be damaged if touched by the tweezer (Figure 4).

Figure 4: Chip and proper chip handling. (A) Yellow dotted line indicates location of the spotted antibodies, or the functional side of the chip. Chip ID is located below line ("58"). Figure also shows proper handling. (B) Demonstrates inappropriate handling of the chip. (C) Nonfunctional side of the chip. Please click here to view a larger version of this figure.
{kind=link}
- Place each pre-scanned chip with the functionalized surface face up into the pre-prepared 24-well plate. The functionalized surface is easily recognized by the distinct numbering, alignment grids, and the three black boxes in the middle. The non-functionalized side is a flat silver uniform surface.
- Use fine point tweezers to ensure that each chip is centered within the well (Figure 5).
NOTE: Centering the chip within the well is critical because if the chip touches the well wall the sample can be wicked off after loading.

Figure 5: Demonstration of proper chip placement in the well. (A) Chips should be set in the middle of the well, with no corners touching the sides of the well. (B) Depiction of improper placement of the chip, where the corners touch the sides of the well. Please click here to view a larger version of this figure.
{kind=link}
3. Loading and incubation of the chip and sample
- Pipette 35 µL of the prepared sample (from step 1) onto the chip.
NOTE: Be careful not to add bubbles or touch the chip with the pipette tip, as it can prevent the even distribution of the sample on the chip or damage the antibody spots on the chip. The sample should spread over the entire chip surface. In the event of the sample wicking off the chip during loading, either add additional sample to the well up to a volume of 250 µL or add incubation solution up to 250 µL and note the changed dilution factor for that sample. - Seal the plate, using the plate film included in the kit to prevent evaporation of the sample.
- Incubate the sample/chip overnight (~16 h) in the sealed plate at room temperature in an area free of vibrations or movement.
NOTE: After the overnight incubation, the user will choose the appropriate Day 2 option for their research question. If only EV size and count are desired, proceed with step 4 (Day 2). If multiple surface marker analysis is desired, skip to step 5 (Day 2).
4. Determining EV size and count (Day 2)
- Add 1 mL of solution A on the side of each well containing a chip, taking care to not directly add the solution on the chip or scratching the chip with the pipette tip.
- Place the plate on an orbital shaker rotating at ~500 rpm for 3 min at room temperature.
NOTE: If the chips rattle on the plate, immediately decrease the speed such that the liquid is swirling but there is no rattling. - Remove 750 µL of liquid. Avoid tilting the plate during liquid removal to prevent accidental drying of the chip.
- Add 750 µL of Solution B using the technique described in step 4.1 and shake at ~500 rpm for 3 min at room temperature.
- Repeat steps 4.3-4.4 twice more for a total of three washes with Solution B.
- At the end of the last wash, remove 750 µL of the solution, leaving 250 µL of Solution B in the well.
- Add 750 µL of double distilled water (ddH2O) and shake at ~500 rpm for 3 min at room temperature.
- Fill a 10 cm Petri dish with 50 mL of ddH2O and transfer one chip at a time from the well into the dish using a tweezer.
NOTE: Take care to transfer the chip horizontally and ensure it does not dry. Up to eight chips can be washed prior to replacing the ddH2O. - In the ddH2O, hold the chip by the edges using a tweezer, and swirl in the dish for three revolutions to remove the debris.
- Remove the chip at a 45° angle out of the water and place the chip on absorbent paper with the chip ID facing up (Figure 6).
NOTE: Chips are now ready to be read. Skip to step 8 for scanning on the SP-IRIS reader.

Figure 6: Correct way to remove the chip out of the ddH2O water at a 45° angle. (A) View from the top and (B) view from the side demonstrating the angle at which to remove the chip. Please click here to view a larger version of this figure.
{kind=link}
5. Preparation of antibody solution (Day 2)
- Add 300 µL of blocking solution per chip into an appropriately sized tube (0.5 to 3 mL).
- Add 0.6 µL of antibody per 300 µL of the blocking solution. Mix gently by tapping the tube and quick spin.
NOTE: Antibodies for human CD9, CD81, and CD63 or murine CD9, CD81, and CD63 are included in the appropriate kits. If other fluorescently conjugated antibodies are desired, determine the optimal staining concentration using a typical titration over a 0.1 to 10 µg/mL range with a constant sample loading as you would for flow cytometry.
6. Determining EV size, count, and phenotyping with immunofluorescent staining
- Add 1 mL of Solution A on the side of each well containing a chip, taking care not to directly add the solution on the chip or scratch the chip with the pipette tip.
- Place the plate on an orbital shaker rotating at ~500 rpm for 3 min at room temperature.
NOTE: If the chips rattle on the plate, immediately decrease the speed such that the liquid is swirling but there is no rattling. - Remove 750 µL of the liquid. Avoid tilting the plate during liquid removal to prevent accidental drying of the chip.
- Add 750 µL of Solution A using the technique described in step 6.1 and shake at ~500 rpm for 3 mins at room temperature.
- Repeat steps 6.3 and 6.4 twice for a total of three washes with Solution A.
- At the end of the last wash, remove 750 µL of the solution, leaving 250 µL of Solution A in the well.
NOTE: If the user desires Cargo Staining, proceed to step 7 at this point. If not, proceed to step 6.7 - Add 250 µL of the Antibody Solution (step 5) to each chip in the well. Cover the plate with foil to protect from light and shake for 1 h on the orbital shaker at room temperature.
- Add 500 µL of Solution A, so the total volume per well is ~1,000 µL.
- Immediately remove 750 µL and add 750 µL of solution A and shake at ~500 rpm on the orbital shaker for 3 min at room temperature.
- Remove 750 µL of the solution and add 750 µL of Solution B and shake at ~500 rpm for 3 mins at room temperature.
- Repeat step 6.10 twice more for a total of three washes.
- Add 750 µL of double distilled water (ddH2O) and shake at ~500 rpm for 3 min at room temperature.
- Fill a 10 cm Petri dish with 50 mL of ddH2O and transfer one chip from the well into the dish using a tweezer.
NOTE: Take care to transfer the chip horizontally and ensure it does not dry. Up to eight chips can be washed prior to exchanging to fresh ddH2O. - In the ddH2O, hold the chip by the edges using a tweezer, and swirl in the dish for three revolutions to remove debris.
- Remove chip at a 45° angle out of the water and place the chip on absorbent paper with the chip ID facing up (Figure 6).
NOTE: Chips are now ready to be read. Skip to step 8 (Data Collection) for instructions on how to set up the chips for scanning on the reader.
7. Optional cargo staining
NOTE: This protocol allows simultaneous labeling of internal and surface markers.
- With 250 µL of Solution A remaining in each well add 250 µL of Solution C into each well.
- Immediately place on the orbital shaker and set to ~200 rpm for precisely 10 min.
NOTE: In this step and step 7.8 below, both timing and slower shaking speed are critical; be sure the speed gives only slow swirling in the wells and use a timer. - After 10 min of incubation with Solution C, add 500 µL of Solution A to each well.
- Immediately remove 750 µL and add 750 µL of Solution A and shake at ~500 rpm on the orbital shaker for 3 min at room temperature.
- Repeat step 7.4 twice more for a total of three washes.
- At the end of the last wash, remove 750 µL of the solution, leaving ~250 µL of the solution in the well with the chip.
- Add 250 µL of Solution D to each well.
- Immediately place on the orbital shaker set to ~200 rpm for precisely 10 min.
- After 10 min of incubation with Solution C, add 500 µL of Solution A to each well.
- Immediately remove 750 µL and add 750 µL of solution A and shake at ~500 rpm on the orbital shaker for 3 min at room temperature.
- Repeat step 7.10 twice for a total of three washes.
- Remove 750 µL of the solution after the last wash and return to step 6.7 above for staining and completing the assay protocol.
8. Data collection
NOTE: The procedure for collecting data from the chips using the ExoView R100 is automated and requires no user inputs. Detailed instructions can be found in the User Guide and corresponding video for loading the chip carrier, or "chuck", and data acquisition17.
- Power on the EV characterization platform using the two power switches on the rear of the instrument, and then start the scanner software by double clicking on the desktop icon.
NOTE: When the scanner software starts, the reader will automatically set to the home screen, and then prompt the user to "Open the door to load chips". - Open the door on the front of the reader by lifting the silver handle. This will eject the stage, allowing the user access to the chuck and set up a new scan in the software.
- Identify the location where the user desires to save the data by clicking on the Save folder and selecting a desired location for the data to be saved.
NOTE: It is often useful to name the save location something insightful like the experiment name, and then create two subfolders, one for pre-scans and one for post-scans. This makes finding and matching the data during analysis easier. - To scan the chips, locate the chipfiles on the control computer.
NOTE: Chipfiles are maps of the antibody spot layout on the chip in use and allow the scanner to know where to scan on each chip. In every kit, there is a USB key with the chipfiles for the chips inside. These should be saved to a location on the control computer upon kit receipt where users can reliably find them. - Load the chips onto the chuck with the number on the chips facing the handle on the chuck, and then from the Chip dropdown menu on the computer, select each chip from the chipfiles list and place it into the appropriate matching location in the virtual chuck in the scanner software. Once complete, an on-screen prompt asking the user to Place the Chuck on Stage will appear.
- Place the loaded chuck on the stage; the magnetic alignment on the chuck will automatically move it to the correct location on the stage, and then click on OK next to Place the Chuck on Stage. The scanner will then begin the automated data collection routine.
NOTE: Data is ready to be analyzed once the software reports the scan status of each chip as successful.
9. Data analysis
- Double click on ExoView Analyzer on the desktop of the control PC. After the software boots, click on the Prescan Data button and select the folder location for the prescan dataset in section 8.3.
- Click on the Postscan Data button and select the folder location for the postscan dataset in section 8.3.
NOTE: If data is properly detected in both the folders, for at least some of the chips, the software will display "X" number of chips detected next to the Chipfile location button. When properly saved, the detected chips should match the number of chips scanned. - Click on Next at the bottom of the data loading area.
- In the meta table dropdown menu, each chip will have a cell. Enter the sample names, dilution factors, and what markers are being stained in each detection channel by clicking in the corresponding box and typing the information in; when completed, click on Next again.
NOTE: This metadata will be saved with the data. - Preform quality control (QC) by clicking on the Disable tab under the QC button in the upper left.
NOTE: This feature allows for specific probes and chips to be turned off for analysis. The software will provide two different warnings regarding the data, one for high counts that can indicate that a particular capture is saturated, and high Coefficient of variation (CV) which identifies when one of the replicates of a capture type is different from the others.- Click on a Warning Spot for high CV, examine the spots that are being called out by the software, and see whether there is obvious physical damage; when the damage is identified, click on the Spot Number to turn that spot off in the analysis.
- Repeat until all the warnings have been evaluated, and then click on Next.
- Preform cutoff analysis by clicking on the Cutoff tab located next to the Disable tab under the QC button in the upper left.
NOTE: This will present the control spot fluorescent response in a chart, and offer the user two settings, a minimum and maximum for each color channel (red/green/blue). Setting the cutoff value for each fluorescent detection channel is the only data adjustment that needs to be made and is relatively simple. Importantly, the final cutoff that is chosen should be consistent across an experiment and as noted should fall within the rules of thumb for 300-400 a.u. in red and green, and 400-700 a.u. for blue in typical experiments; the software has color coded guides for the user for these ranges too.- Click on the Green Color tab to load the data for that channel and display the current cutoffs. Examining the isotype negative control spot one might see detection events with very low fluorescence intensities; in this case, users should typically expect to set the green between 300-400 a.u.
- Increase the minimum for each of the detection channels until the "Avg % Inc" (the average % of the detected particles on the control spot included above the cutoff) under the data display has no warnings as indicated by red or yellow highlighting in that cell.
NOTE: The maximum value does not generally need to be adjusted from the default but can be lowered to filter out bright particles or limit the fluorescent detection to a narrower range. - Click on the Next button.
- Increase the minimum for each of the detection channels until the "Avg % Inc" (the average % of the detected particles on the control spot included above the cutoff) under the data display has no warnings as indicated by red or yellow highlighting in that cell.
- Click on the Red Color tab to load the data for that channel and display the current cutoffs. Again, examining the isotype negative control spot, one might see detection events with very low fluorescence intensities; in this case, users should typically expect to set the red between 300-400 a.u. Follow the same procedures as noted in steps 9.6.1.1-9.6.1.3.
- Click on the Blue Color tab to load the data for that channel and display the current cutoffs. Again, examining the isotype negative control spot, follow the same procedures as noted in steps 9.6.1.1-9.6.1.3.
NOTE: The blue channel is unique in that the antibody in the spot is auto-fluorescent at sufficient levels to require a slightly higher cutoff of 400-700 a.u. even for blank chips.
- Click on the Green Color tab to load the data for that channel and display the current cutoffs. Examining the isotype negative control spot one might see detection events with very low fluorescence intensities; in this case, users should typically expect to set the green between 300-400 a.u.
- Following completion of the Cutoff analysis, click on the Next and the heatmap plot will appear, which will give an overview of the particle binding across the experiment. There are several visualization tools that the user can use to plot the data. These plots and associated raw data can be added to a report by clicking on the Add Plot Report button.
NOTE: The heatmap is the default data display which shows an all samples, all capture view of a particular detection. The default display is the total that quantifies the unique EVs bound to each capture spot. - Click on the Export Report button and select a save location for the report.
NOTE: The report will open in a browser. Find the Filtered Particle List spreadsheet file attached to the report; this is the information to submit to EVTRACK. - Continue adding reports using the different selections of Samples and Capture types in the dropdown menus at the top of the screen. The data displayed is controlled with the selection of channel detection buttons on the top left. Set a particular phenotype/sample selection/capture channel by clicking on each color button to toggle that detection channel off (grayed out) or on (colored).
- Click on the Add Plot to Report button to aggregate the desired plots into a report.
- Click on Export Report and define the save location for the final report.
Results
Figure 7 (left panel) shows a three-color composite image of EVs derived from HEK293 conditioned media bound to the CD63 spot on the chip and stained for CD81, CD63, and CD9 in the following channels green, red, and blue, respectively. Figure 7 (top right panel) is a zoomed-in image that shows each of the captured EVs can display co-localization of one or more colors with varying intensities in each channel. The difference in staining of the captured EVs represe...
Discussion
Current EV characterization methods largely rely on purified EVs, which is restricted by current experimental limitations of EV purification methods9,10,11,12,13.Single-particle interferometric reflectance imaging (SP-IRIS) is an effective technology that can eliminate purification steps required for sample analysis and therefore save time and reduce costs tha...
Disclosures
Clayton Deighan and George Daaboul are employees and shareholders of NanoView Biosciences Inc.
Acknowledgements
This work was sponsored in part by the University of Kansas School of Medicine Research Equipment and Resource Procurement Award Program. PCG, LKC, FD and AR were supported with funds from NIA R21 AG066488-01.
Materials
| Name | Company | Catalog Number | Comments |
| 10-cm sterile Petri dish | Fisher | FB0875712 | |
| 15mL sterile tube | n/a | various | |
| 24-well cell culture plate, flat bottom | Fisher | 08-772-1 | |
| Blocking Solution | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chipfiles | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chips | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chuck | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Corning Easy Grip Disposable Polystyrene Sterile Bottles 250 ml | Fisher | 09-761-4 | |
| Corning Easy Grip Disposable Polystyrene Sterile Bottles 500 ml | Fisher | 09-761-10 | |
| Deionized (DI) water | Fisher | LC267404 | |
| EMS style tweezers with Carbon Fiber tips | Fisher | 50-193-0842 | |
| ExoView Human Tetraspanin Kit | NanoView Biosciences | EV-TETRA-C | Capture for hCD81, hCD9, hCD63, IgG Control + stains for hEV-A (hEV-CD63-647, hEV-CD81-555, hEV-CD9-488) 16 Chips per kit |
| ExoView R100 Imager | NanoView Biosciences | EV-R100 | Interferometric microscope including high specification camera including 3 color fluorescence and label free sizing and counting extracellular vesicles |
| Fluorescently labled huma CD9 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Fluorescently labled human CD63 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Fluorescently labled human CD81 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Incubation Solution | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Orbital shaker or microplate shaker with digital settings capable of shaking at 500 rpm | n/a | various | |
| Plate Seal | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution A | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution B | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution C | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution D | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Square/flat tip tweezer | Fisher | 50-239-62 | |
| Straight strong point Boley style tweezers | Fisher | 16-100-124 | |
| Thermo Scientific Adhesive PCR Plate Seals | Fisher | AB-0558 |
References
- Maas, S. L. N., Breakefield, X. O., Weaver, A. M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends in Cell Biology. 27 (3), 172-188 (2017).
- Shah, R., Patel, T., Freedman, J. E. Circulating extracellular vesicles in human disease. The New England Journal of Medicine. 379 (10), 958-966 (2018).
- Deng, F., Miller, J. A review on protein markers of exosome from different bio-resources and the antibodies used for characterization. Journal of Histotechnology. 42 (4), 226-239 (2019).
- Cohen, O., et al. ’Golden’ exosomes as delivery vehicles to target tumors and overcome intratumoral barriers: in vivo tracking in a model for head and neck cancer. Biomaterials Science. 9 (6), 2103-2114 (2021).
- Han, Y., et al. Overview and update on methods for cargo loading into extracellular vesicles. Processes (Basel). 9 (2), 356 (2021).
- Lee, M., Im, W., Kim, M. Exosomes as a potential messenger unit during heterochronic parabiosis for amelioration of Huntington's disease. Neurobiology of Disease. 155, 105374 (2021).
- Sun, B., et al. Characterization and biomarker analyses of post-COVID-19 complications and neurological manifestations. Cells. 10 (2), 386 (2021).
- Jiang, L., Gu, Y., Du, Y., Liu, J. Exosomes: Diagnostic biomarkers and therapeutic delivery vehicles for cancer. Molecular Pharmaceutics. 16 (8), 3333-3349 (2019).
- Bachurski, D., et al. Extracellular vesicle measurements with nanoparticle tracking analysis - An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. Journal of Extracellular Vesicles. 8 (1), 1596016 (2019).
- Carmicheal, J., et al. Label-free characterization of exosome via surface enhanced Raman spectroscopy for the early detection of pancreatic cancer. Nanomedicine. 16, 88-96 (2019).
- Greening, D. W., Xu, R., Ji, H., Tauro, B. J., Simpson, R. J. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods in Molecular Biology. 1295, 179-209 (2015).
- Wu, Y., Deng, W., Klinke, D. J. Exosomes: improved methods to characterize their morphology, RNA content, and surface protein biomarkers. The Analyst. 140 (19), 6631-6642 (2015).
- van de Vlekkert, D., Qiu, X., Annunziata, I., d'Azzo, A. Isolation and characterization of exosomes from skeletal muscle fibroblasts. Journal of Visualized Experiments: JoVE. (159), (2020).
- Ayala-Mar, S., Donoso-Quezada, J., Gallo-Villanueva, R. C., Perez-Gonzalez, V. H., Gonzalez-Valdez, J. Recent advances and challenges in the recovery and purification of cellular exosomes. Electrophoresis. 40 (23-24), 3036-3049 (2019).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246 (2016).
- Pohler, K. G., et al. Circulating microRNA as candidates for early embryonic viability in cattle. NMolecular Reproduction and Development. 84 (8), 731-743 (2017).
- NanoView Biosciences. ExoView R100 User Guide. v240.4. NanoView Biosciences. , 202 (2021).
- Daaboul, G. G., et al. Enhanced light microscopy visualization of virus particles from Zika virus to filamentous ebolaviruses. PLoS One. 12, 0179728 (2017).
- ET Consortium. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods. 14, 228-232 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved